Workshop 9.8a - Randomized block and repeated measures designs (mixed effects models)
4 August 2011
Basic statistics references
- Logan (2010) - Chpt 12-14
- Quinn & Keough (2002) - Chpt 9-11
Very basic overview of Randomized block
Randomized block
A plant pathologist wanted to examine the effects of two different strengths of tobacco virus on the number of lesions on tobacco leaves. She knew from pilot studies that leaves were inherently very variable in response to the virus. In an attempt to account for this leaf to leaf variability, both treatments were applied to each leaf. Eight individual leaves were divided in half, with half of each leaf inoculated with weak strength virus and the other half inoculated with strong virus. So the leaves were blocks and each treatment was represented once in each block. A completely randomised design would have had 16 leaves, with 8 whole leaves randomly allocated to each treatment.
Download Tobacco data set
| Format of tobacco.csv data files | |||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
 |
Open the tobacco data file.
> tobacco <- read.table("../downloads/data/tobacco.csv", header = T, + sep = ",", strip.white = T) > head(tobacco)
LEAF TREATMENT NUMBER 1 L1 Strong 35.90 2 L1 Weak 25.02 3 L2 Strong 34.12 4 L2 Weak 23.17 5 L3 Strong 35.70 6 L3 Weak 24.12
Since each level of treatment was applied to each leaf, the data represents a randomized block design with leaves as blocks.
The variable LEAF contains a list of Leaf identification numbers and is supposed to represent a factorial blocking variable. However, because the contents of this variable are numbers, R initially treats them as numbers, and therefore considers the variable to be numeric rather than categorical. In order to force R to treat this variable as a factor (categorical) it is necessary to first convert this numeric variable into a factor (HINT).> tobacco$LEAF <- as.factor(tobacco$LEAF)
- H0 Factor A:
- H0 Factor B:
-
Assumption Diagnostic/Risk Minimization I. II. III. IV. - Is the proposed model
balanced?
(Yor N)
> replications(NUMBER ~ LEAF + TREATMENT, data = tobacco)
LEAF TREATMENT
2 8
> !is.list(replications(NUMBER ~ LEAF + TREATMENT, data = tobacco))
[1] TRUE
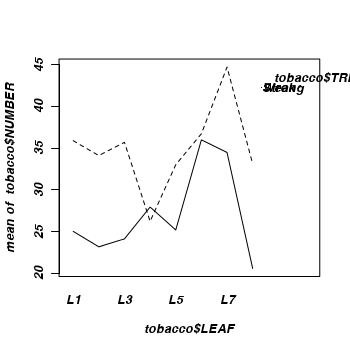
- The simplest method is to plot the number of lesions for each treatment and leaf combination (ie. an
interaction plot
(HINT). Any evidence of an interaction? Note that we cannot formally test for an interaction because we don't have replicates for each treatment-leaf combination!
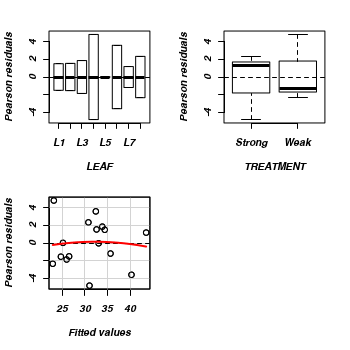
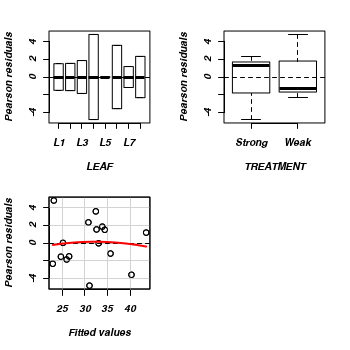
- We can also examine the residual plot from the fitted linear
model. A curvilinear pattern in which the residual values switch from positive to negative
and back again (or visa versa) over the range of predicted values implies that the scale
(magnitude but not polarity) of the main treatment effects differs substantially across the range
of blocks. (HINT). Any evidence of an interaction?
- Perform a Tukey's test for non-additivity evaluated at α = 0.10 or even &alpha = 0.25. This (curvilinear test)
formally tests for the presence of a quadratic trend in the relationship between residuals and
predicted values. As such, it too is only appropriate for simple interactions of scale.
(HINT). Any evidence of an interaction?
- Examine a lattice graphic to determine the consistency of the effect across each of the blocks(HINT).
> library(car) > interaction.plot(tobacco$LEAF, tobacco$TREAT, tobacco$NUMBER)
> residualPlots(lm(NUMBER ~ LEAF + TREATMENT, data = tobacco))
Test stat Pr(>|t|) LEAF NA NA TREATMENT NA NA Tukey test -0.18 0.858
> residualPlots(lm(NUMBER ~ LEAF + TREATMENT, data = tobacco))
Test stat Pr(>|t|) LEAF NA NA TREATMENT NA NA Tukey test -0.18 0.858
> library(alr3) > tukey.nonadd.test(lm(NUMBER ~ LEAF + TREATMENT, data = tobacco))
Test Pvalue -0.1795 0.8575
> library(lattice) > print(barchart(NUMBER ~ TREATMENT | LEAF, data = tobacco))
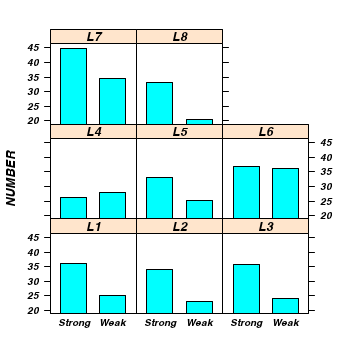
> tobacco.aov <- aov(NUMBER ~ TREATMENT + Error(LEAF), data = tobacco) > summary(tobacco.aov)
Error: LEAF
Df Sum Sq Mean Sq F value Pr(>F)
Residuals 7 292 41.7
Error: Within
Df Sum Sq Mean Sq F value Pr(>F)
TREATMENT 1 248 248.3 17.2 0.0043 **
Residuals 7 101 14.5
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
| Source of variation | df | Mean Sq | F-ratio | P-value |
|---|---|---|---|---|
| LEAF (block) | ||||
| TREAT | ||||
| Residuals |
> library(nlme) > tobacco.lme <- lme(NUMBER ~ TREATMENT, random = ~1 | LEAF, data = tobacco, + method = "ML") > summary(tobacco.lme)
Linear mixed-effects model fit by maximum likelihood
Data: tobacco
AIC BIC logLik
102.5 105.6 -47.25
Random effects:
Formula: ~1 | LEAF
(Intercept) Residual
StdDev: 3.454 3.557
Fixed effects: NUMBER ~ TREATMENT
Value Std.Error DF t-value p-value
(Intercept) 34.94 1.874 7 18.645 0.0000
TREATMENTWeak -7.88 1.901 7 -4.144 0.0043
Correlation:
(Intr)
TREATMENTWeak -0.507
Standardized Within-Group Residuals:
Min Q1 Med Q3 Max
-1.73025 -0.51798 0.01212 0.43724 1.52636
Number of Observations: 16
Number of Groups: 8
> #unique(confint(tobacco.lme,'TREATMENTWeak')[[1]]) > library(gmodels) > ci(tobacco.lme)
Estimate CI lower CI upper Std. Error DF p-value (Intercept) 34.940 30.51 39.371 1.874 7 3.169e-07 TREATMENTWeak -7.879 -12.38 -3.383 1.901 7 4.328e-03
> anova(tobacco.lme)
numDF denDF F-value p-value (Intercept) 1 7 368.5 <.0001 TREATMENT 1 7 17.2 0.0043
> library(lme4) > tobacco.lmer <- lmer(NUMBER ~ TREATMENT + (1 | LEAF), data = tobacco) > summary(tobacco.lmer)
Linear mixed model fit by REML
Formula: NUMBER ~ TREATMENT + (1 | LEAF)
Data: tobacco
AIC BIC logLik deviance REMLdev
96.7 99.8 -44.4 94.5 88.7
Random effects:
Groups Name Variance Std.Dev.
LEAF (Intercept) 13.6 3.69
Residual 14.5 3.80
Number of obs: 16, groups: LEAF, 8
Fixed effects:
Estimate Std. Error t value
(Intercept) 34.94 1.87 18.65
TREATMENTWeak -7.88 1.90 -4.14
Correlation of Fixed Effects:
(Intr)
TREATMENTWk -0.507
> #Calculate the confidence interval for the effect size of the main effect > # of season > library(languageR) > pvals.fnc(tobacco.lmer, withMCMC = FALSE, addPlot = F)
$fixed
Estimate MCMCmean HPD95lower HPD95upper pMCMC Pr(>|t|)
(Intercept) 34.940 34.919 30.83 38.740 0.0001 0.000
TREATMENTWeak -7.879 -7.841 -12.86 -2.231 0.0092 0.001
$random
Groups Name Std.Dev. MCMCmedian MCMCmean HPD95lower HPD95upper
1 LEAF (Intercept) 3.692 1.243 1.440 0.00 3.917
2 Residual 3.803 5.038 5.189 3.08 7.531
> #Examine the effect season via a likelihood ratio test > tobacco.lmer1 <- lmer(NUMBER ~ TREATMENT + (1 | LEAF), data = tobacco, + REML = F) > tobacco.lmer2 <- lmer(NUMBER ~ 1 + (1 | LEAF), data = tobacco, REML = F) > anova(tobacco.lmer1, tobacco.lmer2, test = "F")
Data: tobacco
Models:
tobacco.lmer2: NUMBER ~ 1 + (1 | LEAF)
tobacco.lmer1: NUMBER ~ TREATMENT + (1 | LEAF)
Df AIC BIC logLik Chisq Chi Df Pr(>Chisq)
tobacco.lmer2 3 110 113 -52.2
tobacco.lmer1 4 102 106 -47.2 9.98 1 0.0016 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
> library(ez) > ezANOVA(dv = .(NUMBER), within = .(TREATMENT), wid = .(LEAF), data = tobacco, + type = 3)
$ANOVA
Effect DFn DFd F p p<.05 ges
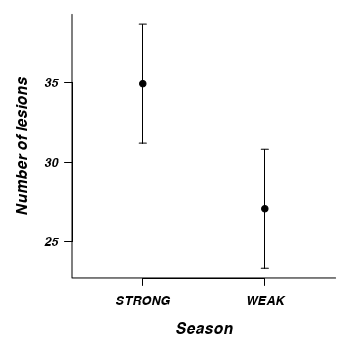
2 TREATMENT 1 7 17.17 0.004328 * 0.387
> newdata <- expand.grid(TREATMENT = c("STRONG", "WEAK"), NUMBER = 0) > mod.mat <- model.matrix(terms(tobacco.lmer), newdata) > newdata$NUMBER = mod.mat %*% fixef(tobacco.lmer) > pvar1 <- (diag(mod.mat %*% tcrossprod(vcov(tobacco.lmer), mod.mat))) > tvar1 <- pvar1 + VarCorr(tobacco.lmer)$LEAF[1] > newdata <- data.frame(newdata, plo = newdata$NUMBER - 2 * sqrt(pvar1), + phi = newdata$NUMBER + 2 * sqrt(pvar1), tlo = newdata$NUMBER - 2 * sqrt(tvar1), + thi = newdata$NUMBER + 2 * sqrt(tvar1)) > par(mar = c(5, 5, 1, 1)) > plot(NUMBER ~ as.numeric(TREATMENT), newdata, type = "n", axes = F, + ann = F, ylim = range(newdata$plo, newdata$phi), xlim = c(0.5, 2.5)) > with(newdata, arrows(as.numeric(TREATMENT), plo, as.numeric(TREATMENT), + phi, ang = 90, length = 0.05, code = 3)) > with(newdata, points(as.numeric(TREATMENT), NUMBER, pch = 21, bg = "black", + col = "black")) > axis(1, at = c(1, 2), lab = newdata$TREATMENT) > axis(2, las = 1) > mtext("Number of lesions", 2, line = 3, cex = 1.25) > mtext("Season", 1, line = 3, cex = 1.25) > box(bty = "l")